CE marking serves as the mandatory passport allowing products to enter and circulate freely across the 30 countries comprising the European Union and European Economic Area. Far more than a simple label, the CE mark represents a manufacturer’s formal declaration that their product complies with all applicable EU health, safety, and environmental protection legislation before being placed on the market. In 2026, with heightened market surveillance enforcement and administrative issues causing 63% of documented compliance failures, understanding the precise CE marking process-from directive identification through technical documentation and conformity assessment-determines whether products gain immediate market access or face customs blocks, recalls, and substantial financial penalties. This comprehensive guide explains exactly what CE marking is, which products require it, how conformity assessment works, what documentation must be prepared, and how manufacturers, importers, and distributors fulfill their legal obligations under EU regulations.
What Is CE Marking?
Definition
CE marking is a mandatory conformity marking for certain products sold within the European Economic Area (EEA), indicating that those products comply with applicable EU legislation covering health, safety, and environmental protection requirements. The CE mark is not a quality certification but rather a legal declaration by the manufacturer that the product meets all relevant European regulatory requirements and can legally be placed on the EU market.
CE Mark Meaning
The letters “CE” stand for “Conformité Européenne,” the French phrase meaning “European Conformity”. By affixing the CE mark to a product, the manufacturer declares on their own responsibility that the product conforms to all applicable EU directives and regulations, has undergone the appropriate conformity assessment procedures, and possesses the required technical documentation.
CE vs. Certification
CE marking is fundamentally a self-declaration of conformity rather than a third-party certification in most cases. While certain high-risk products require assessment by an authorized Notified Body, many products allow manufacturers to self-certify compliance through internal production control processes. This distinguishes CE marking from traditional certification schemes where independent bodies always verify compliance before issuing certificates.
CE vs. Quality Mark
The CE mark addresses only the minimum legal requirements for safety, health, and environmental protection mandated by EU legislation-it does not certify quality, performance, or reliability beyond those regulatory thresholds. Products bearing CE marks meet mandatory legal standards but may vary significantly in quality, durability, and functionality. Quality certifications like ISO standards represent voluntary frameworks that operate independently from the mandatory CE marking system.
EU Legal Basis
CE marking derives its legal authority from the “New Legislative Framework” established by Decision No. 768/2008/EC of the European Parliament and Council, which harmonizes technical product requirements across the EU. Individual product-specific directives and regulations (such as the Low Voltage Directive, Machinery Directive, and Medical Device Regulation) establish the detailed essential requirements that products must meet. These legislations apply uniformly across all EU member states, eliminating technical barriers to trade and enabling the free movement of goods.
Which Products Require CE Marking?
Product Categories
Approximately 20-30 major product categories require CE marking before being placed on the EU market. These categories span diverse industries including electrical equipment, machinery, medical devices, toys, personal protective equipment, construction products, and telecommunications equipment. The specific obligation to bear CE marking depends on whether the product falls within the scope of at least one EU directive or regulation that mandates conformity marking.
EU Directives Overview
Products requiring CE marking include toys, drones, electrical and electronic equipment, pyrotechnic products, recreational craft and watercraft, pressure equipment, gas appliances, batteries, machinery, weighing and measuring equipment, personal protective equipment (PPE), medical devices including diagnostic equipment, radio equipment, construction products, lifts, and equipment intended for use in potentially explosive atmospheres. Each product category is governed by specific EU directives or regulations that establish essential safety requirements and conformity assessment procedures.
Examples
Some products fall under multiple directives simultaneously-for example, a children’s electric toy must satisfy both the Toy Safety Directive and the Low Voltage Directive.
Products That Do NOT Require CE
Many product categories remain outside the scope of CE marking legislation. Products not covered by any EU harmonization directive do not require CE marking, including most food products, pharmaceuticals (which follow separate authorization pathways), cosmetics, unprocessed raw materials, second-hand products sold privately, and many traditional consumer goods like clothing, furniture, books, and household items unless they fall under specific directives. The absence of CE marking requirements does not exempt these products from general safety obligations under the General Product Safety Regulation.
CE Marking Regulations & Directives
Machinery Directive (2006/42/EC)
The Machinery Directive covers machinery, interchangeable equipment, safety components, lifting accessories, chains, ropes, webbing, removable mechanical transmission devices, and partly completed machinery. Manufacturers must conduct risk assessment following EN ISO 12100, ensure compliance with essential health and safety requirements, compile technical documentation, and affix CE marking before placing machinery on the market.
Low Voltage Directive (LVD) (2014/35/EU)
The LVD applies to electrical equipment designed for use with voltage ratings between 50 and 1000 volts AC or 75 and 1500 volts DC. This directive aims to ensure electrical equipment does not endanger persons, domestic animals, or property when properly installed, maintained, and used for its intended purpose. Products must meet safety requirements covering protection against electric shock, fire hazards, mechanical dangers, and other risks.
Electromagnetic Compatibility (EMC) Directive (2014/30/EU)
The EMC Directive requires that electrical and electronic equipment neither generates excessive electromagnetic disturbance that could interfere with other equipment nor be susceptible to electromagnetic interference that would prevent intended operation. All electrical and electronic devices must comply with EMC requirements regardless of whether other directives also apply.
Medical Device Regulation (MDR) (2017/745)
The MDR establishes stringent requirements for medical devices ranging from simple bandages to implantable pacemakers and diagnostic equipment. Medical devices are classified by risk level (Class I, IIa, IIb, III), with higher-risk devices requiring more extensive conformity assessment involving Notified Body certification. The MDR demands clinical evaluation, quality management system implementation, post-market surveillance, and comprehensive technical documentation.
Construction Products Regulation (CPR) (305/2011)
The CPR governs construction products including doors, windows, sealants, structural components, geosynthetics, and building materials. For products covered by harmonized European standards, CE marking is mandatory before market placement. The regulation establishes a Declaration of Performance requirement alongside the Declaration of Conformity.
Toy Safety Directive (2009/48/EC)
The Toy Safety Directive applies to products designed or intended for play by children under 14 years of age. Toys must meet strict requirements for physical and mechanical properties, flammability, chemical composition, electrical properties, hygiene, and radioactivity. Manufacturers must conduct EN 71 testing while also addressing chemical compliance obligations.
Personal Protective Equipment (PPE) Regulation (2016/425)
The PPE Regulation covers equipment designed to be worn or held by a person to protect against health and safety risks, including helmets, masks, safety goggles, protective clothing, and fall protection equipment. PPE is categorized into three risk categories, with higher categories requiring more rigorous conformity assessment involving Notified Bodies.
Radio Equipment Directive (RED) (2014/53/EU)
The RED applies to radio equipment-products that intentionally emit or receive radio waves for communication or radio determination purposes-including radios, Wi-Fi devices, smartphones, IoT devices, and wireless sensors. Products must meet essential requirements for safety, electromagnetic compatibility, and efficient use of radio spectrum.
RoHS Directive (2011/65/EU)
The RoHS Directive restricts the use of certain hazardous substances in electrical and electronic equipment, limiting lead, mercury, cadmium, hexavalent chromium, polybrominated biphenyls, polybrominated diphenyl ethers, and four phthalates. Products must demonstrate compliance through material declarations and, when required, laboratory testing.
Who Is Responsible for CE Marking?
Roles
EU legislation establishes clear responsibilities for different economic operators in the product supply chain. The manufacturer bears primary responsibility for ensuring product compliance, conducting conformity assessment, preparing technical documentation, issuing the Declaration of Conformity, and affixing the CE mark. An importer is any person or legal entity that places products from third countries (non-EU) onto the EU market and must verify that manufacturers have conducted proper conformity assessment, ensure technical documentation and Declarations of Conformity are available, verify CE marking presence, and register products with authorities when required. An authorized representative is appointed by written mandate from manufacturers located outside the EU to act as their EU point of contact, maintain technical documentation and Declarations of Conformity, respond to market surveillance authority requests, and cooperate in corrective actions for non-compliant products. A distributor makes products available on the EU market after they have been placed there by manufacturers or importers, and must verify that products bear CE marking, are accompanied by required documentation in appropriate languages, and comply with applicable requirements.
Responsibility Matrix
The manufacturer always retains ultimate responsibility for conformity assessment even when design, testing, or production is subcontracted. For products manufactured outside the EU, either the manufacturer must appoint an authorized representative within the EU, or the importer assumes manufacturer-level responsibilities.
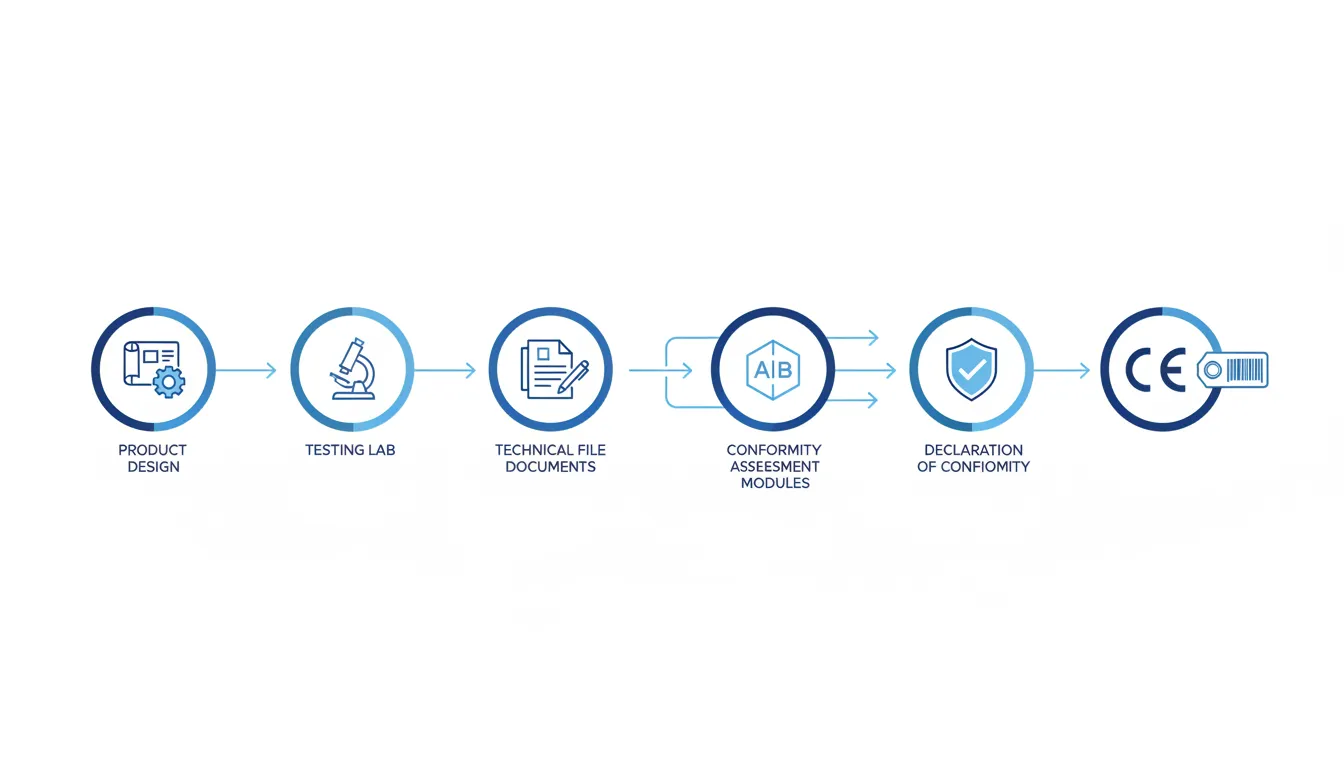
CE Marking Process: Step-by-Step
Identify Applicable Directives
The first critical step requires identifying all EU directives and regulations applicable to your specific product. Many products fall under multiple directives simultaneously-for example, a wireless electric device would be subject to the Low Voltage Directive, EMC Directive, Radio Equipment Directive, and potentially RoHS. Selecting the wrong directive or omitting applicable legislation leads to invalid CE marking and represents the most common technical file error. Manufacturers should analyze product characteristics, intended use, voltage ratings, wireless capabilities, material composition, and user exposure to determine the complete regulatory scope.
Determine Essential Requirements
Each applicable directive establishes “essential requirements”-mandatory health, safety, and environmental protection objectives that products must meet. Essential requirements are stated as outcome-based objectives rather than prescriptive technical specifications. Manufacturers must identify which specific essential requirements apply to their product based on its characteristics, intended use, and foreseeable misuse scenarios. This determination typically requires risk assessment to identify hazards and determine necessary protective measures.
Select Conformity Assessment Procedure
Directives specify one or more conformity assessment procedures (modules) that manufacturers can follow to demonstrate compliance. The appropriate procedure depends on product type, risk classification, and directive requirements. Some products allow pure self-certification through internal production control (Module A), while higher-risk products mandate Notified Body involvement through Type Examination (Module B) followed by production-phase modules. This selection determines whether third-party certification is legally required or optional.
Conduct Testing
Products must undergo testing to verify compliance with essential requirements and, where applied, harmonized standards. Testing may be conducted in the manufacturer’s own facilities for self-certification procedures or by accredited third-party laboratories when required by applicable directives. Tests verify electrical safety, electromagnetic compatibility, mechanical strength, chemical composition, performance characteristics, and other parameters specified by relevant standards. Laboratory testing timelines range from days to weeks depending on test complexity, with wear-and-tear tests for construction products potentially requiring a month.
Compile Technical Documentation
Manufacturers must compile comprehensive technical documentation (also called technical file or technical construction file) demonstrating product conformity with applicable requirements. The technical file must be available as soon as the product is placed on the EU market and maintained for 10 years after the last product is manufactured. Technical documentation includes product description and intended use, conceptual design and manufacturing drawings with explanatory notes, list of applied harmonized standards or descriptions of solutions adopted to meet essential requirements, design calculations and risk assessments, test reports from competent laboratories, copies of conformity documentation for critical components, user instructions and safety information, and the Declaration of Conformity. The documentation must prove that the manufacturer consciously addressed all applicable essential requirements.
Issue Declaration and Affix CE Mark
After completing conformity assessment and compiling technical documentation, the manufacturer issues an EU Declaration of Conformity formally declaring that the product meets all applicable requirements. The Declaration must contain product identification, manufacturer details, references to applicable directives/regulations, harmonized standards applied, description of conformity assessment procedure, Notified Body details if applicable, and authorized signature with date and place. Only after issuing the Declaration of Conformity can the manufacturer affix the CE mark to the product. The CE mark must be visible, legible, and indelible, with a minimum height of 5 mm unless the product size necessitates smaller dimensions.
Conformity Assessment Modules Explained
Modules A–H Overview
The EU employs a modular conformity assessment system with eight modules (A through H) that cover design and production phases. These modules differ in assessment stages, who performs the assessment (manufacturer or Notified Body), and the level of quality system involvement required. Product-specific directives designate which module(s) manufacturers can or must use.
Module A: Internal Production Control (Self-Certification)
Module A allows the manufacturer to independently verify that products meet applicable requirements through internal production control covering both design and manufacturing. The manufacturer ensures conformity, prepares technical documentation, issues the Declaration of Conformity, and affixes CE marking without Notified Body involvement. This represents pure self-certification available for low-risk products.
Module B: EU-Type Examination (Notified Body Required)
Module B requires a Notified Body to examine the technical design of the product and issue an EU-Type Examination Certificate confirming the design meets applicable requirements. Module B addresses only the design stage and must be combined with a production-phase module (C, D, E, or F). The Notified Body reviews technical documentation, examines design adequacy, and may inspect prototypes or type samples. The EU-Type Examination Certificate remains valid as specified by the Notified Body and must be kept available for 10 years.
Module C: Type Conformity Based on Internal Production Control
Module C follows Module B and requires the manufacturer to declare that manufactured products conform to the approved type described in the EU-Type Examination Certificate. The manufacturer internally controls production to ensure conformity against the approved sample. Module C2 adds supervised product checks at random intervals by the Notified Body.
Module D: Production Quality Assurance
Module D follows Module B and requires the manufacturer to operate an ISO 9001-equivalent quality management system covering production, final inspection, and testing. A Notified Body approves the quality system initially and conducts periodic surveillance audits to verify continued compliance. This module emphasizes process control rather than individual product verification.
Module E: Product Quality Assurance
Module E follows Module B and requires a Notified Body-approved quality management system specifically covering final inspection and testing of finished products. The quality system ensures that manufactured products conform to the approved type, with the Notified Body conducting audits to verify system effectiveness.
Module F: Product Verification
Module F follows Module B and requires the Notified Body to examine and test individual products or production batches to verify conformity with the approved type. After verification, the Notified Body issues certificates of conformity for examined products. This module involves product-level third-party inspection rather than system-based assessment.
Module G: Unit Verification
Module G applies to individual products and requires a Notified Body to examine each unit, verify compliance with applicable requirements, and issue individual certificates of conformity. This module applies when products are manufactured individually or in small quantities rather than through series production.
Module H: Full Quality Assurance
Module H requires a comprehensive quality management system covering design, manufacture, final inspection, and testing, all approved and overseen by a Notified Body. Unlike other modules, Module H does not require separate Type Examination (Module B) because the Notified Body assesses both design and production through the quality system. This represents the most comprehensive conformity assessment approach.
Decision Tree Explanation
Decision Process for Selecting Conformity Assessment Module:
- Identify applicable directive(s) – Each directive specifies which modules are available or required for different product types.
- Determine product risk classification – Higher-risk products typically require Notified Body involvement while lower-risk products may allow self-certification.
- Check directive-specific requirements – Some directives mandate specific modules while others offer choices.
- Evaluate production characteristics – Series production suits quality system modules (D, E, H) while individual or small-batch production may use unit verification modules (F, G).
- Assess internal capabilities – Modules requiring quality systems (D, E, H) demand significant internal infrastructure while simpler modules (A, C) require less formalized systems.
- Consider cost and time factors – Notified Body involvement adds cost and time but may provide market credibility advantages.
If the applicable directive allows Module A and the product is low-risk, self-certification through internal production control suffices. If the directive requires Notified Body involvement, start with Module B (Type Examination) then select the production module (C, D, E, or F) that best fits manufacturing processes and internal capabilities. For one-off or small-batch production, Module G (Unit Verification) may be most practical. For manufacturers with established quality systems, Modules D, E, or H leverage existing infrastructure.
When Is CE Certification Required?
Self-Declaration vs. Third-Party Certification
The term “CE certification” can be misleading because CE marking fundamentally represents manufacturer self-declaration of conformity rather than third-party certification in many cases. For numerous product categories, manufacturers can self-certify compliance through Module A (internal production control) without involving any external certification body. However, certain product types and risk classifications legally mandate third-party assessment by authorized Notified Bodies before CE marking can be affixed.
Notified Body Involvement
Each EU directive specifies whether Notified Body involvement is obligatory, optional, or unnecessary for different product types. Notified Bodies must be involved when products fall under directives requiring third-party assessment, when products are classified as high-risk and self-certification is not allowed, or when applicable conformity assessment procedures mandate Notified Body review. Products requiring Notified Body assessment include medical devices with risk factors (Class IIa, IIb, III), higher-category personal protective equipment, pressure equipment above specified thresholds, machinery presenting particular risks, radio equipment using restricted radio spectrum, and construction products under certain assessment systems. When a Notified Body is involved, its four-digit identification number must appear next to the CE mark on the product.
Examples of Products Requiring Notified Body Certification
Medical devices such as surgical instruments, implants, and diagnostic equipment require Notified Body certification. Category II and III personal protective equipment protecting against serious or fatal risks need third-party assessment. Pressure vessels, boilers, and gas appliances above specified pressure or volume thresholds mandate Notified Body involvement. Lifts, cableways designed to carry persons, and certain construction products require third-party certification.
CE Technical Documentation (Technical File)
Technical File Structure
The technical file (also termed technical documentation or technical construction file) represents a comprehensive dossier demonstrating product conformity with all applicable essential requirements. While directives do not prescribe exact formats, the documentation must logically cover product design, manufacture, and operation with sufficient detail to enable authorities to assess compliance. A well-structured technical file typically includes a table of contents, general product description, applicable directives and standards list, risk assessment documentation, design and manufacturing specifications, test reports and certificates, user instructions, and the Declaration of Conformity.
Product Description
Comprehensive product description includes intended use and application scope, technical specifications and operating parameters, product identification (model numbers, variants, serial numbering system), photographs showing all product aspects, and explanation of operating principles. The description must be sufficiently detailed to enable understanding of how the product functions and which essential requirements apply.
Drawings & Schematics
Technical drawings communicate product design and construction, including general assembly drawings showing overall product configuration, detailed engineering drawings of critical components, electrical circuit diagrams and wiring schematics for electronic products, control system layouts, and exploded views showing assembly relationships. Drawings must be professional, dimensioned, and include explanatory notes necessary for understanding.
Bill of Materials (BOM)
A complete bill of materials lists all components, sub-assemblies, and materials used in product construction. The BOM should identify component part numbers, supplier details, material specifications, quantities, and applicable conformity documentation for purchased components. For products subject to chemical restrictions (RoHS, REACH), the BOM must enable traceability of material composition.
Risk Assessment
Risk assessment represents a mandatory element for most products, identifying hazards, assessing risks, and documenting protective measures implemented to achieve acceptable risk levels. Risk assessment should follow recognized methodologies such as EN ISO 12100 for machinery. The assessment must consider intended use, reasonably foreseeable misuse, all lifecycle phases (installation, operation, maintenance, disposal), and identify applicable essential requirements based on identified hazards. Absence of proper risk assessment invalidates CE marking.
Test Reports
Test reports from competent laboratories or manufacturer testing facilities demonstrate that products meet applicable technical requirements. Reports should reference specific test standards, describe test specimens and conditions, present detailed results with measurements and observations, and conclude whether products pass or fail requirements. Test reports must come from accredited laboratories when third-party testing is mandatory.
Standards Applied
Documentation must list all harmonized European standards applied during product development and conformity assessment. When harmonized standards are applied in full, products benefit from a presumption of conformity with covered essential requirements. If harmonized standards are applied only partially or not at all, documentation must describe alternative solutions implemented to meet essential requirements. Omitting standard references represents a frequent Declaration of Conformity error.
Component Declarations
For products incorporating purchased components, sub-assemblies, or modules, the technical file should include conformity documentation from suppliers. This includes Declarations of Conformity or Declarations of Incorporation for sub-assemblies, material compliance certificates for RoHS and REACH, safety data sheets for chemicals, and test reports for critical components.
EU Declaration of Conformity
What It Is
The EU Declaration of Conformity (DoC) is a formal legal document in which the manufacturer declares, on their sole responsibility, that the product complies with all applicable EU legislation. The Declaration represents the culmination of the conformity assessment process and serves as the manufacturer’s guarantee that essential requirements have been met. Without a valid Declaration of Conformity, products cannot legally bear CE marking or be placed on the EU market.
Required Contents
A valid Declaration of Conformity must contain specific mandatory information:
- Product identification – Model number, type designation, article number, batch or serial number enabling precise identification
- Manufacturer’s name and complete address – Legal name and physical address where the manufacturer can be contacted
- Authorized representative details (if applicable) – Name and address of any appointed EU representative
- Statement of responsibility – Declaration that conformity is issued under the sole responsibility of the manufacturer
- Product description – Sufficient description to enable correlation with the physical product
- References to applicable EU legislation – Complete list of directives and/or regulations with which the product complies, including official numbers
- Harmonized standards applied – References to harmonized European standards used, including standard numbers and publication dates
- Notified Body information (when applicable) – Name, identification number, and certificate reference of any involved Notified Body
- Authorized person’s name, position, signature, date, and place – Signature of person authorized to bind the manufacturer legally
Incorrect or incomplete Declarations of Conformity legally invalidate CE marking. Common errors include missing directive references, incomplete standard listings, incorrect manufacturer details, missing product model identification, and absence of authorized signature.
When Declaration Must Accompany Product
For most product categories, the Declaration of Conformity must be available to market surveillance authorities immediately upon request but does not need to physically accompany each product. However, certain directives explicitly require that each product be accompanied by its Declaration of Conformity, including machinery, equipment for potentially explosive atmospheres, radio and telecommunications terminal equipment, measuring instruments, recreational craft, lifts, high-speed and conventional rail systems, and constituents of the European Air Traffic Management network. For these products, the Declaration must ship with each unit or be immediately accessible.
Template Explanation
While no single official template exists, Declarations of Conformity typically follow a standardized format. The document begins with the title “EU Declaration of Conformity” or “EC Declaration of Conformity”. The opening section identifies the manufacturer and authorized representative. The body declares that the described product complies with listed directives/regulations and standards. When a Notified Body was involved, their details and certificate references appear. The declaration concludes with place, date, authorized person’s name and function, and signature. One consolidated Declaration can address multiple applicable directives rather than requiring separate declarations for each.
Affixing the CE Mark Correctly
Placement Rules
The CE marking must be affixed to the product itself or, when not possible due to the product’s nature, to its packaging or accompanying documents. The mark should be affixed in a location that is visible, accessible, and will remain legible throughout the product’s expected lifetime. It can appear on the back or underside of products, provided it remains easily accessible to all parties. For small products where the minimum size requirement cannot be met on the product itself, the CE mark must appear on packaging or, if that is impossible, in accompanying documentation.
Size Requirements
The CE mark must have a minimum height of 5 mm measured vertically. This minimum dimension may be waived for small devices or components where physical size constraints prevent compliance. When enlarging or reducing the CE mark, the proportions between the two letters and the specified construction must remain identical. Letter spacing, line thickness, and the ratio between the letters “C” and “E” must not be distorted.
Visibility & Legibility
The CE mark must be visible, legible, and indelible. Visible means the mark can be seen without disassembling the product or removing permanent fixtures. Legible means the mark is clear, readable, and maintains required proportions. Indelible means the mark is permanently affixed and cannot be removed without leaving obvious traces-it must withstand normal handling, environmental conditions, and product use throughout its expected lifetime. The CE marking can take different forms regarding color (monochrome or contrasting with background), solid or hollow lettering, but the initials must remain clearly visible and recognizable.
Labeling Rules
When affixed, the CE marking must be accompanied by specific information:
- Product identification – Type, batch, or serial number detailed on the product or label
- Manufacturer name and address – Legal name and postal address where the manufacturer can be contacted
- Importer name and address (for imported products) – Contact details for the importer who placed the product on the EU market
- Notified Body identification number (when applicable) – Four-digit number of the Notified Body that performed conformity assessment, positioned immediately after the CE mark
Incorrect or missing labeling information represents a leading cause of CE marking non-compliance identified during market surveillance. Missing manufacturer or importer identification, absent product type/serial numbers, and incorrect or missing Notified Body numbers are among the most frequent administrative failures.
Digital CE Marking
While traditional physical affixing remains the norm, some directives allow digital CE marking in specific circumstances. Digital CE marking may appear on electronic displays for products where physical marking is impractical, provided the mark meets visibility, legibility, and permanence requirements throughout product operation. The specific conditions for digital CE marking depend on applicable directive provisions.
CE Marking for Importers & Non-EU Manufacturers
Importer Obligations
Importers play a critical compliance role when bringing products from non-EU countries onto the European market. Before placing products on the market, importers must verify that the manufacturer has conducted appropriate conformity assessment procedures, ensure the product bears CE marking with correct Notified Body identification (if applicable), verify that the product is accompanied by required documentation in a language easily understood by consumers and users in the destination country, and ensure the product is accompanied by instructions and safety information. Importers must verify CE marking is visible, legible, and indelible on the product. If an importer has reason to believe a product is non-compliant, they must not place it on the market until conformity is ensured.
Authorized Representative Requirements
Manufacturers located outside the European Union must appoint an authorized representative established within the EU by written mandate. The authorized representative serves as the point of contact for market surveillance authorities and maintains specific documentation. The representative’s responsibilities include keeping the EU Declaration of Conformity and technical documentation available for inspection by national authorities for 10 years after the last product is placed on the market, providing authorities with all information and documentation necessary to demonstrate product conformity, and cooperating with competent authorities on any actions taken to eliminate risks posed by products covered by their mandate.
Documentation Access
Importers and authorized representatives must ensure immediate availability of technical files and Declarations of Conformity upon request from market surveillance authorities. Under Regulation (EU) 2019/1020 on market surveillance, all products must have an appointed “responsible party” or economic operator before placement on the European market. This party must verify CE marking, register as the EU point of contact, ensure availability of technical files and Declarations of Conformity, cooperate with market surveillance authorities, and inform authorities when there is reason to believe a product presents a risk.
Market Surveillance Context
With heightened enforcement under the Market Surveillance Regulation, importers and authorized representatives face increased scrutiny. Administrative issues cause the majority of non-compliance cases, with CE marking, product identification, manufacturer identification, importer identification, and Declaration of Conformity among the most frequent failure points. Technical documentation issues caused 63% of non-compliance cases in documented market surveillance activities, with missing general product descriptions and incomplete lists of applied harmonized standards particularly common.
CE Marking Costs & Timeframes
Cost Factors
CE marking costs vary dramatically based on multiple factors. Product type fundamentally determines costs-simple products require minimal testing while complex machinery demands extensive conformity assessment. Testing requirements represent major expenses, with laboratory fees based on time investment and test complexity. Single test procedures may require several days of equipment time, while specialized tests like wear-and-tear assessments for construction products can take a month. Notified Body involvement adds substantial costs when required, as third-party assessment, certification, and ongoing surveillance demand payment of Notified Body fees. Documentation preparation requires significant time investment from qualified personnel, with even straightforward technical files requiring at least a full day of work and complex products demanding weeks or months. Consultant engagement adds hourly or project-based fees when internal expertise is insufficient.
Typical Cost Ranges
CE certification costs easily exceed $1,500 even for relatively simple products when accounting for testing, documentation, and professional time. Low-complexity products like simple toys or textiles might incur costs in the $2,000-$5,000 range. Medium-complexity electrical devices typically cost $5,000-$15,000 for testing, documentation, and compliance verification. High-complexity products like machinery, pressure equipment, or medical devices can cost $15,000-$100,000 or more depending on testing scope, Notified Body involvement, and documentation complexity. Medical devices under the MDR, particularly higher risk classifications, may require substantially higher investments due to clinical evaluation, quality system certification, and extensive technical documentation requirements.
Timeframe Factors
CE marking timelines depend on product complexity, applicable directives, testing requirements, Notified Body involvement (when required), preparedness of technical documentation, and whether initial testing reveals non-conformities requiring corrective action. Product complexity directly affects documentation time and testing duration. Directive requirements vary in stringency and documentation demands. Testing duration ranges from days for simple electrical safety tests to weeks or months for complex or specialized assessments. Notified Body review adds weeks or months to the process when third-party certification is required. Documentation preparation takes longer when starting from scratch versus updating existing files.
Typical Timelines
On average, CE marking timelines range from 2 weeks to 6 months. Simple products like basic toys or textiles typically require 2-4 weeks. Electrical devices usually need 4-8 weeks for testing and documentation. Machinery and pressure vessels typically require 2-6 months due to complexity and testing requirements. Products requiring Notified Body involvement generally need 3-6 months or longer depending on Notified Body workload and review complexity. Medical devices under the MDR can take substantially longer-often 12-24 months for higher risk classifications due to clinical evaluation, quality system audits, and comprehensive technical documentation requirements. These timeframes assume technical files are ready, required information is available, and testing yields positive results. Incomplete documentation, failed tests requiring redesign, or incorrect initial applications can significantly extend timelines.
CE Marking Lifecycle Compliance
Design Stage
Compliance begins during conceptual design when fundamental product architecture, material selections, and safety features are established. Design-stage compliance activities include identifying all applicable directives and essential requirements, conducting preliminary risk assessment to identify hazards and necessary protective measures, selecting harmonized standards to be applied during development, integrating safety features into product design rather than adding them later, documenting design decisions and their compliance rationale, and planning conformity assessment approach and testing strategy. Design-for-compliance principles prevent costly redesigns discovered during testing or documentation preparation.
Production Phase
Manufacturing must ensure that series production maintains conformity with the design verified during conformity assessment. Production-phase compliance includes implementing quality control procedures appropriate to the selected conformity assessment module, maintaining production documentation that enables traceability, conducting in-process inspections and final product testing, managing changes to materials, components, or processes that could affect compliance, and maintaining consistency with the approved type (when Module B Type Examination was performed). For modules D, E, and H requiring quality management systems, manufacturers must maintain Notified Body-approved systems and submit to periodic surveillance audits.
Market Placement
Placing products on the EU market requires specific documentation and marking activities. Market placement compliance includes affixing CE marking correctly with required visibility, legibility, and permanence, ensuring products carry required identification (type, batch, serial numbers), providing user instructions and safety information in appropriate languages, issuing EU Declaration of Conformity, making technical documentation available for market surveillance authorities, and registering products with authorities when applicable directives require registration. For imported products, importers must verify these requirements are met before distribution.
Post-Market Monitoring
Compliance obligations continue throughout the product’s commercial lifetime. Post-market compliance activities include monitoring product performance and safety incidents in the field, maintaining systems to receive and investigate customer complaints, reporting serious incidents to authorities when required by applicable directives, implementing corrective actions including field safety notices or recalls when necessary, updating technical documentation when products are modified, monitoring regulatory changes that could affect continued compliance, and maintaining technical files for required retention periods (typically 10 years after last product manufactured). Medical device manufacturers face particularly stringent post-market surveillance obligations including periodic safety update reports.
Updates & Modifications
When products are modified, manufacturers must assess whether changes affect conformity with applicable requirements. Significant changes may require repeating conformity assessment, updating technical documentation, revising the Declaration of Conformity, and potentially engaging Notified Bodies for reassessment. Minor changes that provably do not affect compliance may require only documentation updates. Change management procedures should evaluate technical impact, regulatory implications, documentation requirements, and Notified Body notification obligations.
Risks of Non-Compliance
Fines & Penalties
Non-compliant CE marking exposes manufacturers, importers, and distributors to enforcement actions by national market surveillance authorities. Penalties vary by member state but can include substantial administrative fines calculated based on violation severity and economic advantage gained through non-compliance. Repeated or intentional violations incur higher penalties. Beyond direct fines, non-compliance generates costs for corrective actions, additional testing, documentation remediation, and legal fees.
Market Bans
Products failing to meet CE marking requirements face prohibition orders preventing market placement and requiring withdrawal of already-distributed products. Market surveillance authorities can ban products from the EU market entirely when serious non-compliance is identified. Unlike fines that represent one-time costs, market bans eliminate revenue from affected products until compliance is achieved. For products already distributed through retail channels, bans require coordinating withdrawal with all parties in the supply chain.
Product Recalls
Serious non-compliance, particularly involving safety risks, can mandate product recalls requiring notification to purchasers, collection of distributed products, refunds or replacements, and disposal or remediation of non-compliant products. Recall costs include reverse logistics, customer communications, refund processing, potential liability claims, and brand damage. Recalls generate ongoing compliance monitoring obligations and heightened regulatory scrutiny.
Customs Blocking
Products lacking valid CE marking or required documentation face detention at EU customs entry points. Customs authorities verify CE marking presence, proper product identification, and Declaration of Conformity availability before clearing imports. Non-compliant products are refused entry and must be returned to origin, destroyed, or brought into compliance before importation. Customs blocking creates supply chain disruptions, product delivery delays, and potential contract breaches with customers.
Liability Exposure
Products placed on the market without valid CE marking expose manufacturers, importers, and distributors to product liability claims if safety issues arise. Non-compliance with essential safety requirements increases liability risk and may invalidate insurance coverage. Economic operators who knowingly place non-compliant products on the market face potential criminal liability in some member states. Authorized representatives who fail to cooperate with authorities or maintain required documentation can be held liable within their mandate scope.
Reputational Damage
CE marking failures damage business reputation with customers, distributors, and regulatory authorities. Public disclosure of non-compliance through market surveillance databases and safety alerts permanently associates companies with compliance failures. Distributors and retailers may refuse to carry products from manufacturers with compliance histories, while B2B customers increasingly conduct supplier compliance audits that reveal past issues.
CE Marking vs. Other Certifications
CE Marking (EU)
CE marking represents mandatory regulatory compliance for specified product categories sold in the 30-country European Union and European Economic Area. It addresses essential health, safety, and environmental requirements established by EU legislation. CE marking is not a quality certification but rather a legal prerequisite for market access. Products bearing CE marks may be placed on the market in all EU/EEA member states without additional national approvals for the aspects covered by harmonized directives.
UKCA Marking (United Kingdom)
UKCA (UK Conformity Assessed) marking replaced CE marking for the Great Britain market (England, Wales, Scotland) following Brexit. UKCA covers most products previously requiring CE marking under EU directives, with requirements generally mirroring EU essential requirements. Northern Ireland continues following EU rules under the Northern Ireland Protocol, meaning CE marking remains valid there. During transition periods, CE marking remained accepted for certain product categories in Great Britain, but UKCA represents the permanent GB conformity marking.
FCC Certification (United States)
FCC (Federal Communications Commission) certification applies to electronic devices capable of emitting radio frequency energy sold in the United States. FCC rules cover both intentional radiators (devices designed to emit RF signals) and unintentional radiators (devices that may generate RF emissions as a byproduct). Unlike CE marking’s self-declaration option, FCC certification requires testing by FCC-accredited laboratories and submission of technical documentation to the FCC. FCC certification addresses only radio frequency emissions and does not cover the broader safety, EMC, and environmental requirements encompassed by CE marking.
UL Certification (United States/Canada)
UL (Underwriters Laboratories) certification represents voluntary third-party safety certification rather than mandatory regulatory compliance. While not legally required for most products, UL certification demonstrates compliance with recognized safety standards and is often demanded by retailers, insurance companies, and large purchasers. UL focuses specifically on product safety-electrical safety, fire hazards, mechanical safety-without addressing electromagnetic compatibility, radio frequency emissions, or environmental requirements. Unlike CE marking, UL certification always involves third-party testing and certification by UL or other Nationally Recognized Testing Laboratories (NRTLs).
Products entering multiple markets require compliance with all applicable certifications-for example, a wireless electronic device sold in both the EU and US would need CE marking, FCC certification, and potentially UL certification.
Common CE Marking Mistakes
Wrong Directives Identified
Selecting incorrect or incomplete applicable directives represents the most fundamental CE marking error. Common mistakes include evaluating electrical products only under the Low Voltage Directive while ignoring mandatory EMC requirements, performing toy safety tests under EN 71 but neglecting chemical compliance obligations under other directives, ignoring Radio Equipment Directive requirements for IoT and wireless devices, and failing to recognize that single products often fall under multiple simultaneous directives. Wrong legislation leads to wrong technical files and invalid CE marking. Manufacturers must systematically analyze all product characteristics-voltage ratings, wireless capabilities, intended users, material composition-to identify the complete regulatory scope.
Missing or Invalid Tests
Test-related errors include missing required tests entirely, conducting tests to wrong standards or outdated standard versions, testing non-representative samples that differ from production units, using non-accredited laboratories when accreditation is required, and failing to test all product variants covered by a single Declaration of Conformity. Test reports must reference specific harmonized standards or other technical specifications, describe test specimens accurately, present complete results with measurements, and conclude clearly on pass/fail status. Technical documentation that omits test reports or includes incomplete test evidence cannot demonstrate conformity.
Incomplete Technical File
Technical file deficiencies represent a leading cause of market surveillance non-compliance findings. Common technical file mistakes include missing product drawings and schematics (dimensions, layouts, circuit diagrams, bill of materials, material specifications, assembly drawings), absent or inadequate risk assessment (no assessment conducted, using non-standard methods instead of EN ISO 12100, listing only theoretical risks, ignoring foreseeable misuse), incomplete or non-compliant user manuals (missing required languages, absent safety warnings, missing installation/maintenance/disposal instructions), missing general product description, incomplete list of applied harmonized standards, and absent copies of component supplier declarations. The absence of a proper risk assessment invalidates CE marking regardless of other documentation quality. Product drawings are among the first documents requested during inspections, and their absence immediately signals inadequate compliance.
Incorrect Declaration of Conformity
Declaration of Conformity errors include missing or incorrect directive/regulation references, incomplete standard listings, incorrect or incomplete manufacturer details, missing product model identification, absence of authorized signature, incorrect Notified Body references (when applicable), and missing date or place of issuance. If the Declaration of Conformity is incorrect, the CE marking is legally invalid. The Declaration must precisely identify which directives/regulations the product complies with, list all harmonized standards applied (with numbers and versions), identify the manufacturer and authorized representative completely and accurately, describe the product unambiguously, and bear the signature of a person with authority to legally bind the manufacturer.
Incorrect CE Mark Affixing
Physical marking errors identified during market surveillance include CE marks smaller than the required 5 mm minimum height, distorted proportions where letter spacing or ratios are incorrect, insufficient visibility or legibility, non-permanent marking that can be easily removed or wears off quickly, incorrect placement making the mark inaccessible, missing Notified Body identification number when required, and absent or incorrect accompanying information (manufacturer name, product identification). Incorrect CE mark affixing can render otherwise compliant products non-compliant and subject to enforcement action.
Future of CE Marking
Digital Product Passport Integration
The European Union’s Digital Product Passport (DPP) initiative will transform how compliance information is accessed and verified. DPPs create machine-readable digital records linked to physical products through QR codes, NFC tags, or other digital identifiers. These passports provide comprehensive product information including manufacturer identification, material composition, performance characteristics, repair information, end-of-life instructions, and compliance status. CE marking documentation will increasingly integrate with DPP systems, enabling real-time regulatory verification, automated customs clearance, and consumer transparency. Products in electronics, textiles, and batteries sectors face DPP requirements beginning in 2026-2027. This digitalization will require manufacturers to structure technical documentation in formats enabling digital accessibility rather than relying solely on PDF files and paper archives.
Sustainability Directives Expansion
The EU’s Ecodesign for Sustainable Products Regulation (ESPR) extends compliance obligations beyond traditional safety and environmental requirements into comprehensive sustainability performance. Future CE marking will encompass material efficiency requirements, product durability and reparability standards, recycled content obligations, carbon footprint declarations, and circular economy design principles. These sustainability requirements will be phased in through product-specific implementing regulations, with iron and steel products among the first categories in 2026. Technical files will expand to include lifecycle assessments, recycling protocols, material traceability documentation, and end-of-life management plans. This evolution transforms CE marking from primarily safety-focused compliance into comprehensive product stewardship encompassing environmental performance throughout the entire product lifecycle.
Smart Compliance & Automation
Technological advances enable increasingly automated compliance management. Future compliance systems will employ artificial intelligence to monitor regulatory changes across all EU directives, automatically identify applicable requirements for specific products, flag design or material choices that could create compliance issues, and generate compliant technical documentation from structured product data. Integration between product lifecycle management (PLM) systems, compliance software, and regulatory intelligence platforms will embed compliance verification into design and engineering workflows rather than treating it as a separate downstream activity. Market surveillance authorities will increasingly employ automated systems that verify compliance data against digital product identities, enabling real-time enforcement and reducing reliance on physical inspections. Manufacturers investing in digital compliance infrastructure will achieve faster time-to-market, reduced compliance costs, and greater confidence in regulatory adherence compared to those maintaining manual processes.
Frequently Asked Questions
How to CE mark a product?
CE marking requires six core steps: (1) identify all applicable EU directives and regulations, (2) determine essential requirements your product must meet, (3) select and execute the appropriate conformity assessment procedure (Module A through H depending on product type), (4) conduct required testing to verify compliance with applicable standards, (5) compile comprehensive technical documentation demonstrating conformity, and (6) issue an EU Declaration of Conformity and affix the CE mark to the product. The specific requirements, testing needs, and whether Notified Body involvement is required depend entirely on your product category and applicable legislation.
Can I self-certify for CE marking?
Self-certification through Module A (internal production control) is permitted for many low- and medium-risk product categories where applicable directives do not mandate Notified Body involvement. However, certain products legally require third-party assessment by authorized Notified Bodies, including higher-risk medical devices, Category II and III personal protective equipment, pressure equipment above specified thresholds, and machinery presenting particular hazards. You must check the specific conformity assessment requirements in all applicable directives to determine whether self-certification is allowed or third-party assessment is mandatory.
What products require CE marking?
Approximately 20-30 product categories require CE marking, including toys, electrical and electronic equipment, machinery, medical devices, personal protective equipment, construction products, gas appliances, pressure equipment, radio equipment, measuring instruments, recreational craft, lifts, and equipment for explosive atmospheres. Whether your specific product requires CE marking depends on whether it falls within the scope of at least one EU directive or regulation mandating conformity marking. Products not covered by any CE marking directive are exempt, even though they must still comply with general safety obligations.
What is CE certification?
The term “CE certification” is technically imprecise because CE marking fundamentally represents manufacturer self-declaration of conformity rather than third-party certification in many cases. However, the phrase is commonly used to describe the entire CE marking process including conformity assessment, technical documentation compilation, and marking affixation. For products requiring Notified Body involvement, “CE certification” more accurately describes the process since a third-party certification body formally assesses and certifies compliance. Regardless of terminology, CE marking represents a legal prerequisite for placing covered products on the EU market.
How long does CE marking take?
CE marking timelines range from 2 weeks to 6 months on average, depending on product complexity and applicable directives. Simple products like basic toys or textiles typically require 2-4 weeks. Standard electrical devices need 4-8 weeks for testing and documentation. Complex machinery and pressure vessels usually require 2-6 months. Products requiring Notified Body assessment generally need 3-6 months or longer. Medical devices under the MDR can take 12-24 months for higher risk classifications due to clinical evaluation, quality system certification, and comprehensive technical documentation requirements. These timeframes assume technical files are ready and testing yields positive results-incomplete documentation or failed tests significantly extend timelines.
Do startups need CE marking?
Yes, if startups manufacture or import products falling under applicable CE marking directives, they must comply with all CE marking requirements before placing products on the EU market regardless of company size or age. Regulatory authorities make no exceptions for startups, small businesses, or first-time manufacturers. Startups often face particular challenges meeting CE marking requirements due to limited resources, lack of regulatory expertise, and pressure to reach market quickly. However, non-compliance creates severe risks including customs blocking, market bans, fines, and inability to sell through established distribution channels. Startups should integrate compliance into product development from the earliest design stages rather than treating it as a final hurdle before launch.
Is CE marking valid forever?
CE marking remains valid as long as the product continues to comply with applicable EU legislation and the underlying design, materials, or manufacturing processes do not change. However, several factors can affect continued validity: (1) regulatory changes-when directives are revised or new requirements are introduced, existing products may need reassessment, (2) product modifications-significant changes to design, materials, or intended use may invalidate existing CE marking and require new conformity assessment, (3) Notified Body certificates-when Notified Bodies are involved, their certificates have specified validity periods (often 3-5 years for some products) and require renewal, and (4) quality system surveillance-products certified under quality system modules (D, E, H) require ongoing Notified Body surveillance audits. Manufacturers must maintain technical documentation for 10 years after the last product is manufactured, even though the CE marking itself has no formal expiration date.
CE marking represents far more than a compliance formality-it determines whether products can access the €16+ trillion European economic zone, influences operational workflows from design through post-market surveillance, and directly affects competitive positioning in global markets. Companies that approach CE marking strategically-integrating regulatory requirements into product development, maintaining robust technical documentation systems, and understanding conformity assessment options-achieve faster market access, avoid costly redesigns, and build sustainable compliance foundations for product portfolios. As the EU expands CE marking into sustainability dimensions through Digital Product Passports and the Ecodesign for Sustainable Products Regulation, manufacturers who invest in sophisticated compliance infrastructure today position themselves for competitive advantages tomorrow.