Entering the United States market requires navigating one of the world’s most rigorous regulatory environments: the Food and Drug Administration (FDA). The FDA regulates over 20 cents of every dollar spent by US consumers, covering everything from lasagna and lipstick to lasers and life-saving heart valves.
For manufacturers and importers, “FDA Compliance” is not a single certificate. It is a continuous lifecycle of registration, quality control, labeling, and post-market surveillance. A common misconception is that all products need “FDA Approval.” In reality, most require Registration or Clearance, and knowing the difference is the first step to avoiding detention at the US border.
This guide provides a comprehensive operational framework for bringing FDA-regulated products to market in 2026, incorporating the latest updates from MoCRA (cosmetics), DSCSA (pharma traceability), and LDT (lab-developed tests) final rules.
What Is FDA Compliance?
FDA Compliance means adhering to the laws enforced by the FDA under the Federal Food, Drug, and Cosmetic Act (FD&C Act) and related regulations in Title 21 of the Code of Federal Regulations (21 CFR).
Scope and Authority
The FDA is a federal law enforcement agency. Its authority allows it to:
- Inspect facilities (foreign and domestic).
- Seize non-compliant products.
- Refuse entry at the border (Import Alerts).
- Prosecute companies and individuals criminally.
Products Regulated by FDA
The FDA does not regulate all products (e.g., it does not regulate toys or car seats). It specifically oversees:
- Foods: Packaged foods, produce, seafood, dietary supplements, bottled water.
- Drugs: Prescription (Rx) and Over-the-Counter (OTC) medications.
- Medical Devices: From tongue depressors (Class I) to pacemakers (Class III).
- Cosmetics: Perfumes, makeup, moisturizers, shampoos.
- Biologics: Vaccines, blood products, gene therapies.
- Radiation-Emitting Electronics: Microwaves, X-ray machines, laser pointers.
- Tobacco Products: Vapes, cigarettes, cigars.
- Animal & Veterinary: Livestock feed, pet food, veterinary drugs.
FDA Approval vs. Registration vs. Listing
This is the most critical distinction for new entrants. “FDA Approved” is a specific legal status that applies to very few products. Misusing this term is “misbranding” and illegal.
| Term | Meaning | Applies To |
|---|---|---|
| FDA Registered | The facility has notified FDA it exists. Does NOT imply endorsement. | Food facilities, Medical Device mfrs, Drug mfrs, Cosmetic facilities (New MoCRA rule). |
| FDA Listed | The product is entered into the FDA database. Does NOT imply passing a test. | Medical devices, Drugs, Cosmetics. |
| FDA Cleared | FDA has reviewed a 510(k) submission and agreed the device is “substantially equivalent” to one already on the market. | Most Class II Medical Devices. |
| FDA Approved | FDA has reviewed rigorous clinical data and determined the benefits outweigh the risks. | Class III Medical Devices (PMA), New Drugs (NDA), Biologics. |
- Key Takeaway: You generally cannot say a dietary supplement or cosmetic is “FDA Approved.” You can say the facility is “FDA Registered.”
FDA Risk Classification Framework
The regulatory burden scales with risk.
Medical Devices (Class I, II, III)
- Class I (Low Risk): Bandages, sunglasses, manual toothbrushes. Most are exempt from pre-market review; just register and list.
- Class II (Moderate Risk): Syringes, powered wheelchairs, pregnancy tests. Usually require 510(k) Clearance.
- Class III (High Risk): Implants, life-sustaining devices. Require Premarket Approval (PMA) with clinical trials.
Drugs (OTC vs. Rx)
- OTC Monograph: If your drug (e.g., sunscreen, fluoride toothpaste) follows a public “recipe book” (Monograph), you don’t need approval. You just register and list.
- New Drug Application (NDA): Novel drugs require years of clinical trials and formal approval.
Foods
- FSMA (Food Safety Modernization Act): Focuses on preventing contamination (HACCP/HARPC plans) rather than “approving” recipes.
Core FDA Regulations Framework
To operate legally, you must align with these four pillars:
- 21 CFR (Code of Federal Regulations): The detailed rulebook.
- Part 111: GMP for Dietary Supplements.
- Part 820: Quality System Regulation (QSR) for Devices.
- Part 210/211: GMP for Drugs.
- cGMP (Current Good Manufacturing Practices): The “how-to” for manufacturing. You must prove you can make the product consistently and safely, not just test the final batch.
- Labeling Regulations: Strict rules on font sizes, “Nutrition Facts” panels, and ingredients lists.
- Reporting: Mandatory reporting of “Adverse Events” (injuries/deaths) linked to your product.
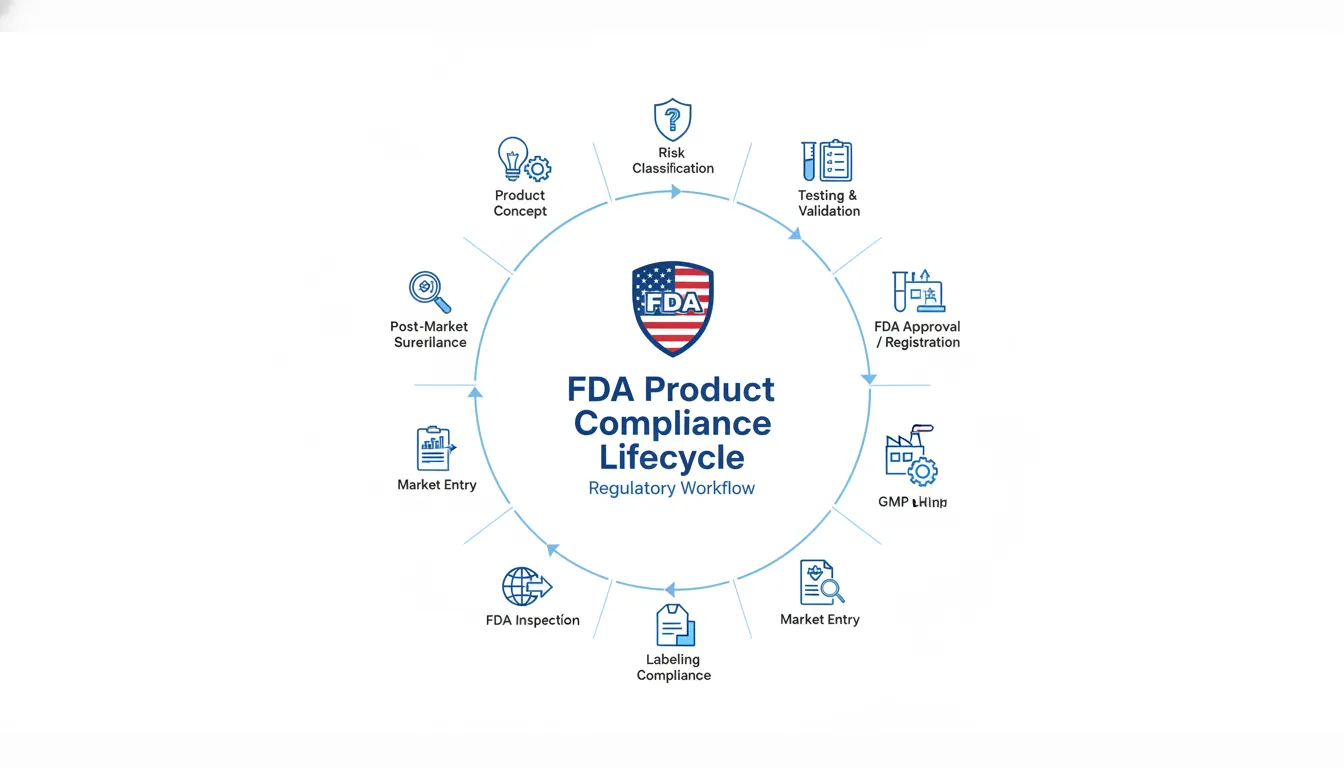
FDA Compliance Lifecycle
Compliance is not a one-time event. It follows this lifecycle:
Phase 1: Pre-Market
- Classification: Determine if it’s a drug, device, or cosmetic.
- Facility Registration: Obtain an FEI (FDA Establishment Identifier) and pay user fees (for devices/drugs).
- US Agent: Foreign facilities must appoint a US Agent physically located in the US to receive legal documents.
Phase 2: Market Entry
- Submission: File 510(k), cosmetic listing (MoCRA), or drug listing.
- Labeling Review: Ensure labels comply before printing.
- Import: File “Prior Notice” with CBP for foods; ensure Registration Number is on commercial invoices.
Phase 3: Post-Market
- Surveillance: Monitor complaints.
- Inspection: Be ready for FDA investigators to inspect your factory (even overseas).
- Renewal: Biennial registration renewal (typically Oct-Dec of even-numbered years).
Registration & Listing (2026 Updates)
Cosmetics (MoCRA)
Under the Modernization of Cosmetics Regulation Act (MoCRA), cosmetic compliance has shifted significantly:
- Mandatory Facility Registration: Manufacturers must register their facility (Form 5066).
- Product Listing: Each product formula must be listed (Form 5067) with ingredients and contact info.
- Deadline: Existing products should have been registered by mid-2024; new products must register within 60 days of marketing.
Medical Devices & Drugs
- Establishment Registration: Must be renewed annually (Oct 1 – Dec 31).
- Fees: Device manufacturers pay an annual fee (~$9,000 in 2026). Drug/Food facility registration is free (but US Agent costs apply).
FDA Documentation Architecture
You must maintain a Technical File (Device Master Record or Batch Record) proving compliance.
- Product Specifications: Exact formula, drawings, and component list.
- Manufacturing Procedures (SOPs): Step-by-step instructions for production.
- Testing Records: Proof that this specific batch met specifications.
- Labeling Copies: The exact artwork used.
- Supply Chain Records: Who did you buy ingredients from? (FSVP for food importers).
Labeling & Claims Compliance
Labeling errors are the #1 cause of import detentions.
The “Intended Use” Trap
FDA classifies products based on claims, not ingredients.
- Example: Charcoal powder.
- Claim: “Cleans teeth” → Cosmetic.
- Claim: “Whitens teeth” → Drug/Cosmetic (Structure/Function).
- Claim: “Cures gum disease” → New Drug (Illegal without NDA approval).
Mandatory Elements
- Statement of Identity: What is it? (e.g., “Dietary Supplement”).
- Net Quantity: Metric and Imperial units.
- Ingredients List: In descending order by weight.
- Manufacturer/Distributor: Name and Address.
- “Rx Only” symbol for prescription devices/drugs.
Quality Systems (GMP / QSR)
You cannot “inspect quality into” a product; you must build it in.
- Medical Devices (21 CFR 820 / ISO 13485): You need a Quality Management System (QMS) covering Design Controls, CAPA (Corrective and Preventive Action), and Supplier Management.
- Dietary Supplements (21 CFR 111): You must test 100% of finished batches for identity, purity, strength, and composition, unless you have a statistical exemption.
- Foods (21 CFR 117): Hazard Analysis and Risk-Based Preventive Controls (HARPC). You must have a “PCQI” (Preventive Controls Qualified Individual) oversee the plan.
FDA Inspections
FDA inspections are “guilty until proven innocent.” The investigator arrives unannounced (for domestic) or scheduled (for foreign).
Types of Inspections
- Pre-Approval Inspection (PAI): Before approving a new drug/Class III device.
- Routine Surveillance: Every 2-3 years based on risk.
- For-Cause: Triggered by a whistle-blower, recall, or customer complaints.
The Outcome: Form 483
If investigators find violations, they issue a Form 483 listing them.
- Response: You have 15 business days to respond with a corrective action plan. Failure to do so leads to a Warning Letter.
Enforcement & Violations
- Warning Letter: Public notification of serious violations. This destroys your reputation and often causes customers (like Amazon/Walmart) to drop you.
- Import Alert: The “Red List.” Your products are detained automatically at the border without physical examination. You must prove compliance to get off the list (expensive and slow).
- Seizure/Injunction: US Marshals seize goods; court orders stop production.
Importing FDA-Regulated Products
The Importer of Record is legally responsible for compliance.
The “Prior Notice” Rule
For food/supplements, you must file Prior Notice with FDA before the goods arrive.
Foreign Supplier Verification Program (FSVP)
If you import food/supplements, you cannot just trust the foreign factory. You (the US importer) must have a qualified individual audit the foreign supplier and keep records proving they meet US safety standards.
Common FDA Compliance Mistakes
- “It works in Europe”: CE Marking data is not automatically accepted by FDA. They are totally different systems.
- Labeling Claims: Using words like “treats,” “cures,” “prevents,” or “heals” on a supplement or cosmetic. This makes it an unapproved drug.
- Registration vs. Approval: Marketing a “Class I” device as “FDA Approved.” (It’s only registered).
- Ignoring CAPA: Failing to document how you fixed a problem. FDA cares more about how you fix mistakes than the mistake itself.
FAQ
Do all products need FDA approval?
No. Only New Drugs and Class III Medical Devices typically need strict “approval.” Most others need “Registration” (listing yourself in the database) and adherence to GMP/labeling rules.
How do I register with FDA?
You typically use the FDA Unified Registration and Listing Systems (FURLS). It’s an online portal.
What is GMP?
Good Manufacturing Practices. It’s a quality system ensuring products are consistently produced and controlled. It requires SOPs, training, and testing.
Can I sell without approval?
For many low-risk products (cosmetics, most foods, Class I devices), yes-as long as you are registered, listed, and compliant with labeling/GMPs. You don’t need to wait for a certificate.
What is a US Agent?
A person or company physically in the US who acts as a liaison for a foreign facility. They must differ from your commercial importer. They are available 24/7 for emergency communications.